Mit Etuvetidigen Autotemcel wurde Ende 2025 in den USA erstmals eine Ex-vivo-Gentherapie beim seltenen Wiskott-Aldrich-Syndrom (WAS) zugelassen. Auch die europäische Arzneimittelbehörde EMA hat die Zulassung empfohlen. Die Behandlung ist eine Option für Betroffene ohne passenden Stammzellspender.
Das WAS ist ein X-chromosomal-rezessiv vererbter Immundefekt bei bis etwa 1 : 100 000 Lebendgeborenen, der sich durch Mikrothrombozytopenie mit Blutungsneigung, Ekzeme, rezidivierende Infekte, Autoimmunerkrankungen und ein erhöhtes Lymphomrisiko äußert. Kinder mit der potenziell lebensbedrohlichen Erkrankung können schon kurz nach der Geburt mit Petechien auffallen, oder mit einem Ekzem, das einer atopischen Dermatitis ähnelt. Im Verlauf drohen unter anderem gastrointestinale oder intrakranielle Blutungen [1,2].
Ursache sind Mutationen im WAS-Gen. Das von ihm kodierte WAS-Protein (WASP) reguliert das Aktin-Zytoskelett in hämatopoetischen Zellen. Die Krankheitsschwere korreliert mit dem Mangel an funktionsfähigem WASP und hängt vom Genotyp ab, der zunehmend als prognostischer Biomarker dient. Mehrere hundert pathogene Varianten sind bekannt. Wer gar kein WASP produziert, hat ohne kurative Therapie eine Lebenserwartung unter 15 Jahren [2,3,4].
Zwar kann leichter Erkrankten eine supportive Behandlung und Prophylaxe, etwa mit Thrombozyten-Transfusionen, Immunglobulin-Gaben und Antibiotika genügen, doch die Erkrankung kann sich jederzeit verschlechtern. Deshalb befürworten Expertinnen und Experten inzwischen, allen jungen Erkrankten eine potenziell kurative Therapie anzubieten [1,5].
Stammzellspende am besten im Alter unter 5
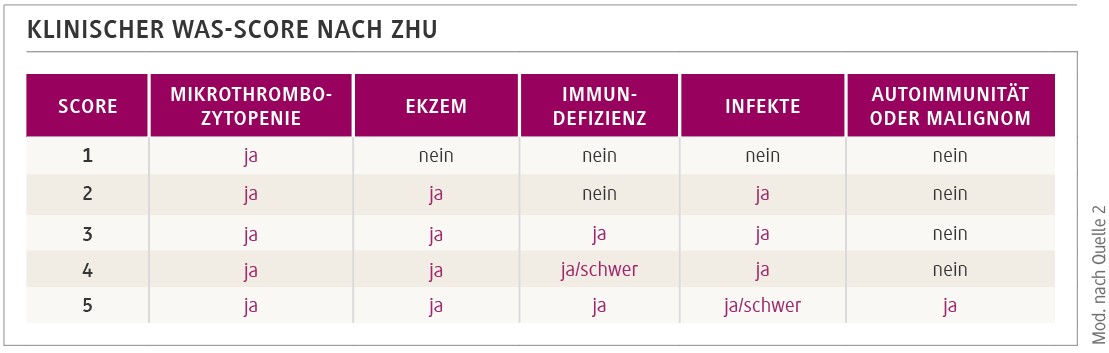
Goldstandard und spätestens bei einem Zhu-Score > 3 (Abb.) indiziert ist die allogene hämatopoetische Stammzelltransplantation (HSZT). Dank verbesserter Technologien und Konditionierungs-Regime beträgt die 3-Jahres-Überlebensrate inzwischen nahezu 90 %. Selbst wenn sich ein HLA-identischer Spender findet, bleiben aber typische Risiken, wie Transplantatabstoßungen, Graft-versus-Host-Disease (GvHD) oder Autoimmun-Komplikationen, vor allem bei nicht verwandten Spendern und bei Empfängern, die älter sind als 5 Jahre [5,6].
Als Alternative experimentieren Forscherinnen und Forscher deshalb seit Jahren mit Gentherapien. Ansätze mit γ-Retrovirus-Vektoren scheiterten daran, dass die meisten Patienten bzw. Patientinnen infolge einer Insertionsmutagenese Leukämien und Myelodysplasien entwickelten. Bei mit selbstinaktivierenden Long Terminal Repeats ausgestatteten Drittgenerations-Lentivirus(LV)-Vektoren, wie in dem jetzt in den USA zugelassenen Etuvetidigen Autotemcel, passiert das offenbar nicht. Bislang hat kein mit einer LV-Gentherapie Behandelter Malignome entwickelt, und das bei Follow-ups bis zu 13 Jahren [2,3].
Gentherapie nutzt eigene Stammzellen aus peripherem Blut
Vorgesehen ist die neue Gentherapie für WAS-Erkrankte ab 6 Monaten mit HSZT-Indikation, aber ohne geeigneten Spender. Mit dem Vektor wird im Labor eine funktionierende WAS-Genkopie in die aus eigenem peripherem Blut gewonnenen hämatopoetischen Stammzellen eingebracht. Nach Konditionierung des Empfängers bekommt er seine genetisch modifizierten Stammzellen in einer Sitzung reinfundiert. Sie wandern ins Knochenmark, vermehren sich dort und reifen zu gesunden Blutplättchen und Immunzellen [7].
Infektionen und Blutungen rückläufig
US-Zulassung und EMA-Empfehlung beruhen auf 2 offenen, einarmigen Studien und einem erweiterten Zugangsprogramm. Bei den insgesamt 27 Kindern mit schwerem WAS verbesserten sich morbiditäts- und mortalitätsrelevante Manifestationen signifikant. Die jährliche Rate schwerer Infektionen sank von 2 im Jahr vor Gentherapie auf 0,15 in den ersten 2 Jahren danach und im Folgejahr weiter auf 0,12. Gab es im Jahr vor der Gentherapie noch 2 moderate bis schwere Blutungen, waren es im Jahr 2–3 nur noch 0,16 pro Jahr [6,7,8].
Unerwünschte Wirkungen wie Blutungen an der Katheterstelle oder Infektionen entstanden vornehmlich im Kontext Infusion selbst oder der Vorbehandlung und Konditionierung mit Rituximab sowie dosisreduziertem Busulfan/Fludarabin [7].
Autoimmun-Flares bei vorbestehenden Autoimmunmanifestationen bleiben noch eine Herausforderung. Zugleich macht Gen-Editing die Therapien immer präziser. Forschende gehen davon aus, dass künftige Strategien optimierte Übertragungssysteme, auch Lipid-Nanopartikel, kombinieren, um eine sichere, wirksame und zelllinienadaptierte WASP-Expression zu ermöglichen [2].
Die erste in den USA zugelassene Gentherapie bietet also eine wirksame und risikoärmere kurative Alternative zur HSZT für WAS-Erkrankte ohne geeigneten Spender. Als autologes Verfahren umgeht sie das Risiko einer GvHD und ist weniger toxisch, weil die Konditionierung keine komplette Myeloablation erfordert. Ekzeme und schwere Blutungen verschwinden meist, schwere Infektionen werden seltener. Viele der Betroffenen können auf Immunglobulin-Gaben verzichten.