

Angeborene Cholestase-Syndrome werden traditionell der Pädiatrie zugeordnet, treten jedoch auch im Erwachsenenalter in Erscheinung – bei Persistenz von Erkrankungen im Kindesalter, nach Lebertransplantation oder als späte, triggerabhängige Erstmanifestation („adult-onset“).
Die Entstehung hereditärer Cholestasen kann durch eine primäre oder sekundäre Störung der Gallensäuresekretion begünstigt werden [1]. Das kann zur Akkumulation von Gallensäuren in den Hepatozyten führen sowie zum Rücktransport von Gallensäuren ins sinusoidale Blut. Entsprechend sind intrahepatische Cholestasen mit Gallensäuresekretionsstörung durch hohe Serumgallensäuren, erhöhte Werte für Alaninaminotransferase (ALT) und Aspartataminotransferase (AST) sowie normwertige oder im Vergleich zur Cholestase niedrige Werte für Gamma-Glutamyltransferase (GGT) gekennzeichnet [1].
Die neue EASL-Leitlinie
Die Leitlinien der EASL (European Association for the Study of the Liver) von 2024 empfehlen bei unklarer intrahepatischer Cholestase eine frühzeitige, phänotypgeleitete genetische Abklärung sowie ein strukturiertes Management des cholestatischen Pruritus [2]. Im Zentrum stehen Defekte der kanalikulären Gallensekretion und -zusammensetzung. Klinisch relevant sind [2,3]:
ABCB4-assoziierte Erkrankungen (MDR3-Defekt) sind im Erwachsenenalter besonders bedeutsam, da heterozygote wie biallelische Varianten ein Kontinuum von niedrigphospholipidassoziierter Cholelithiasis (LPAC) über rezidivierende Choledocholithiasis/Cholangitis bis zur chronischen cholestatischen Hepatopathie bedingen können [4,5]. Pathobiologisch führt eine reduzierte Phospholipidsekretion in die Galle zu erhöhter Membran-/Cholangiotoxizität und biliärer Verletzung [5].
Klinische Präsentation im Erwachsenenalter
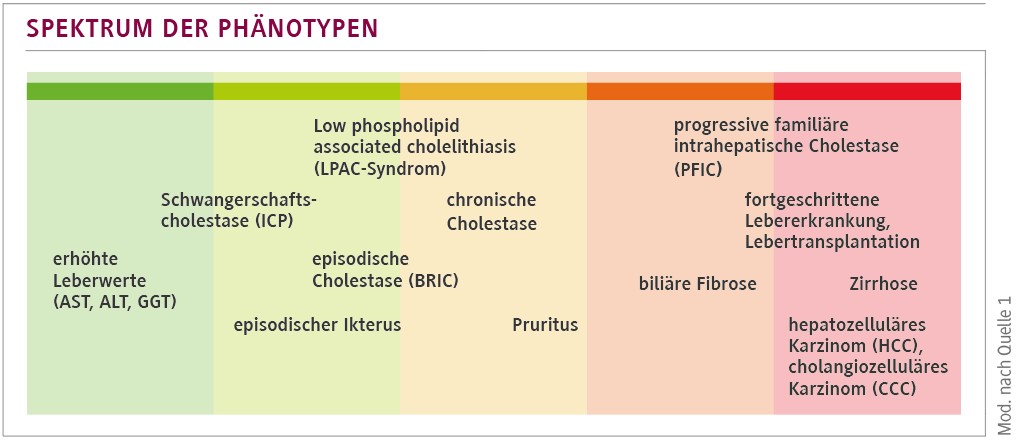
Leitsymptome sind cholestatischer Pruritus, Ikterus und Fatigue. Hinzukommen Steatorrhö/Malabsorption sowie Komplikationen chronischer Cholestase (Abb.). Typische Trigger sind Infekte, Medikamente (inklusive hormoneller Kontrazeptiva), Stress und Schwangerschaft [3,6]. In einer Kohorte mit Erkrankungsbeginn > 18 Jahre wurden Varianten in ABCB4, ABCB11 und ATP8B1 bei 28,4 % der getesteten Patientinnen und Patienten gefunden [6].
Auch bei „ungeklärter“ Cholestase können genetische Varianten häufig relevant sein: In einer skandinavischen Studie wurden bei mindestens 27–54 % genetische Ursachen als sicher bzw. beitragend bewertet, insbesondere bei Schwangerschaftsassoziation oder Persistenz pathologischer Werte post partum [3]. Biochemisch ist die Unterscheidung „low-GGT“ vs. „high-GGT“ diagnostisch hilfreich, ersetzt jedoch keine molekulare Sicherung [2,3].
Diagnostik
Zunächst müssen häufigere Ursachen abgegrenzt werden (extrahepatische Obstruktion, DILI*, PBC **/ PSC***, Virushepatitiden). Bei persistierender ungeklärter Cholestase, rezidivierenden Episoden, frühem Beginn im Leben, auffälliger Familienanamnese oder Cholestase-Ereignissen in der Schwangerschaft sollte ein genetisches Panel (Cholestase-/Transporter-Gene) veranlasst werden [2]. Bildgebung dient primär der Ausschlussdiagnostik. Leberhistologie kann die Phänotypzuordnung stützen und die Interpretation genetischer Varianten flankieren [2,6].
Therapie und Langzeitmanagement
Therapieziele sind Symptomkontrolle (v. a. Pruritus), Prävention der Progression sowie Trigger- und Sondersituationsmanagement (Schwangerschaft, perioperativ). Ursodesoxycholsäure (UDCA) wurde ursprünglich zur Therapie bei Gallensteinerkrankung zugelassen und später bei verschiedenen cholestatischen Krankheiten getestet [1]. UDCA akkumuliert in der Galle und verdünnt dadurch toxische endogene Gallensäuren. Zudem fördert UDCA die hepatozytäre Gallensekretion sowie die biliäre Bicarbonatsekretion und wirkt antiapoptotisch. UDCA ist bei ABCB4-assoziierten Phänotypen häufig wirksam.
Der cholestatische Pruritus sollte stufenweise leitlinienbasiert behandelt werden (u. a. Gallensäurebinder, Rifampicin, Opioidantagonisten; jeweils unter Leberwert- und Interaktionsmonitoring) [2]. In refraktären Verläufen kommen in spezialisierten Zentren u. a. Plasmapherese oder endoskopische Drainageverfahren in Betracht [2].
Bei progredienter Erkrankung (z. B. PFIC-Phänotyp, fortgeschrittene ABCB4-Erkrankung) bleibt die Lebertransplantation die definitive Option. Parallel sind genetische Beratung, Familienscreening und bei Frauen die vorausschauende Schwangerschaftsberatung essenziell, um Rezidive, maternale Komplikationen und fetale Risiken früh zu adressieren [1,2].
*drug-induced liver injury (DILI), arzneimittelinduzierter Leberschaden
**primär biliäre Cholangitis (PBC)
***primär sklerosierende Cholangitis (PSC)

Bildnachweis: privat
