
Ein vielgesichtiges Syndrom, das scheinbar nicht miteinander verknüpfte und im Erwachsenenalter beginnende Manifestationen verbindet, hat an Profil gewonnen. Es gibt Scoring-Systeme, um das VEXAS-Syndrom zu (er-)fassen. Die Therapieoptionen stellen jedoch noch nicht zufrieden.
Das Akronym VEXAS steht für: Vakuolen, Ubiquitin-aktivierendes E1-Enzym, X-chromosomal bedingt, Autoinflammatorisch und Somatisch [1]. Das so bezeichnete, erstmals 2020 beschriebene Syndrom wird durch eine somatische Partial-loss-of-function-Mutation im Ubiquitin-like-modifier-activating-enzyme-1(UBA1)-Gen in hämatopoetischen Stamm- und Progenitorzellen ausgelöst. Die Folge sind (Auto-)Inflammtion, Zytopenien sowie ein erhöhtes Risiko für myelodysplastische Neoplasien und multiples Myelom.
Man sollte das VEXAS-Syndrom kennen
Die Frequenz der pathogenen Mutation liegt insgesamt bei rund 1 : 13 600. Bei Männern über 50 Jahre, die häufiger betroffen sind, da sie nur ein X-Chromosom haben, sogar bei 1 : 4 300 [2]. Dies bedeute, dass in Berlin etwa 270 Menschen mit einem VEXAS-Syndrom lebten, rechnete Prof. Dr. med. Matthias Goebeler (Würzburg) vor. Er geht von einer hohen Dunkelziffer aus.
Die 5-Jahres-Überlebensrate bei Menschen mit VEXAS-Syndrom liege nach dem Auftreten der ersten Manifestationen bei etwa 80 %, erklärte der Experte. In 4–50 % der Fälle entwickelt sich ein myelodysplastisches Syndrom (MDS), wobei (prä-)maligne Dysplasien von entzündlichen Veränderungen oft schwer zu differenzieren sind. Ob ein MDS die Prognose des VEXAS-Syndroms verschlechtert, ist bisher unklar. Auch das multiple Myelom tritt beim VEXAS-Syndrom vermehrt auf, obwohl Lymphozyten nicht mutiert sind.
Red Flags
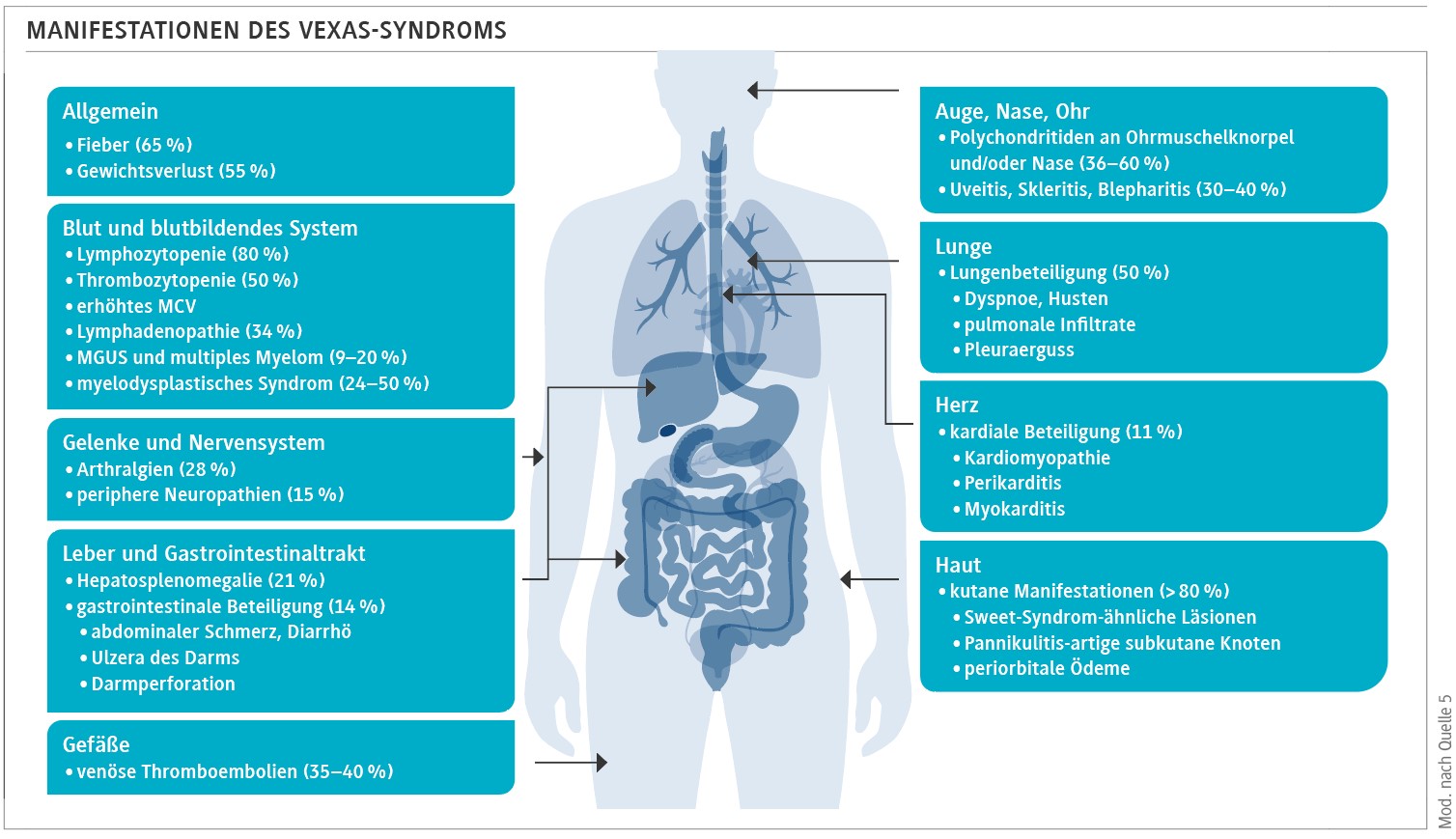
Das VEXAS-Syndrom kann viele Organsysteme beeinträchtigen, über 80 % der Manifestationen betreffen jedoch die Haut. Daneben ist Fieber ein häufiges Symptom, außerdem Gewichtsverlust und Veränderungen im Blutbild und fast immer eine makrozytäre Anämie. Auch Polychondritiden an Ohrmuschelknorpel und in der Nase sind häufig. Da vermehrt venöse Thromboembolien in der Anamnese beobachtet wurden, sollte hiernach gefragt werden, riet Goebeler [3].
Die mit dem ersten Buchstaben des Akronyms genannten Vakuolen treten in hämatopoetischen Vorläuferzellen auf, gelegentlich auch in (unreifen) Neutrophilen und Monozyten des peripheren Bluts. Die Häufigkeit liegt jedoch nur zwischen 3 und 15 %, sodass das Fehlen dieser Vakuolen ein VEXAS-Syndrom nicht ausschließt.
Bei wem der Genstatus erhoben werden sollte
Die VEXAS-Läsionen zeigen eine große morphologische Variabilität auf der Haut (Abb.). Um Patienten und Patientinnen für eine zielführende Abklärung des UBA1-Genstatus zu identifizieren, wurde in Japan ein einfacher Score mit folgenden Kriterien entwickelt:
Das Vorliegen einer makrozytären Anämie wird mit 2 Punkten bewertet, jedes andere Kriterium mit 1 Punkt. Bei einem Score von 6 Punkten gilt VEXAS als sehr wahrscheinlich, bei 5 Punkten als wahrscheinlich, bei 3 als möglich und darunter als unwahrscheinlich. Alle Getesteten mit einem Score von ≥ 3 Punkten sollten einer genetischen Abklärung zugeführt werden. Wie Goebeler berichtete, habe man in der japanischen Studie bei einem Score von 6 Punkten die Mutation immer nachweisen können [4]. Ein zweites Scoring-System aus den USA sei „etwas sperriger im Alltag“ [5].
Therapiestrategien und -optionen
Therapeutisch komme ein antiinflammatorischer Ansatz infrage, alternativ könne der mutierte Klon bekämpft oder eine Kombination aus beiden Ansätzen angewendet werden, erklärte Goebeler.
Die einzige kurative Therapie ist eine allogene hämatopoetische Stammzelltransplantation, die jedoch selbst mit einer hohen Morbidität verbunden sei. Als innovative Strategien werden pharmakologische Aktivatoren für UPA1 sowie des Proteasoms diskutiert. Bisher fehlen zudem konsentierte Therapie-Leitlinien für das VEXAS-Syndrom.
Ubiquitinierung
Folgen des teilweisen UBA-Funktionsverlusts
Bei einer Ubiquitinierung wird Ubiquitin über eine 3-stufige Enzym-Kaskade (E1, E2, E3) auf andere Proteine übertragen [6]. Die Ubiquitinierung verändert Funktion und Eigenschaften der Zielproteine, häufig führt sie zum Abbau durch das Proteasom. Ubiquitinierungen sind Teil vieler zellulärer Prozesse wie Transkription und Translation, Signaltransduktion und DNA-Reparatur.
Der mutationsbedingte Funktionsverlust beim VEXAS-Syndrom betrifft nur eines der Enzyme auf Ebene 1 der Ubiquitinierungskaskade (Infokasten): UBA1 – nicht aber UBA2. Auf der Ebene E2 erhalten daher Enzyme, die normalerweise durch UBA1 ubiquitiniert würden, keine Markierung. Die UBA2-abhängige Ubiquitinierung anderer Enzyme verläuft hingegen normal. Dies verändert auch die Ubiquitinierung auf der Ebene E3 und es kommt zu einer Imbalance der markierten Proteine. Im Zytoplasma akkumulieren überflüssige und falsch gefaltete Proteine. Dies aktiviert die „Unfolded Protein Response“ (UPR) sowie weitere Signalkaskaden, die über Typ-1-Interferon und den NF-κB-Signalweg die Autoinflammation befördern [7].
Vortrag „Autoinflammatorische Syndrome und das VEXAS-Syndrom: Neue Erkenntnisse zu Pathomechanismen und Therapie“
